Genetic Disorders
A genetic disorder is a disease caused by a different form of a gene called a variation, or an alteration of a gene called a mutation. Many diseases have a genetic aspect. Some, including many cancers, are caused by a mutation in a gene or group of genes in a person's cells. These mutations can occur randomly or because of an environmental exposure such as cigarette smoke.
Other genetic disorders are inherited. A mutated gene is passed down through a family and each generation of children can inherit the gene that causes the disease. Still other genetic disorders are due to problems with the number of packages of genes called chromosomes. In Down syndrome, for example, there is an extra copy of chromosome 21.
If you know that you have a genetic problem in your family, you can have genetic testing to see if your baby could be affected.
Down Syndrome
Down syndrome is set of mental and physical symptoms that result from having an extra copy of chromosome 21. Even though people with Down syndrome may have some physical and mental features in common, symptoms of Down syndrome can range from mild to severe. Usually, mental development and physical development are slower in people with Down syndrome than in those without it.
People with the syndrome may also have other health problems. They may be born with heart disease. They may have dementia. They may have hearing problems and problems with the intestines, eyes, thyroid and skeleton.
The chance of having a baby with Down syndrome increases as a woman gets older. Down syndrome cannot be cured. However, many people with Down syndrome live productive lives well into adulthood.
People with the syndrome may also have other health problems. They may be born with heart disease. They may have dementia. They may have hearing problems and problems with the intestines, eyes, thyroid and skeleton.
The chance of having a baby with Down syndrome increases as a woman gets older. Down syndrome cannot be cured. However, many people with Down syndrome live productive lives well into adulthood.
Marfan Syndrome
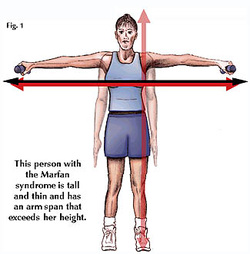
Marfan syndrome is a disorder of connective tissue, the tissue that strengthens the body's structures.
Disorders of connective tissue affect the skeletal system, cardiovascular system, eyes, and skin.
Causes, incidence, and risk factorsMarfan syndrome is caused by defects in a gene called fibrillin-1. Fibrillin-1 plays an important role as the building block for connective tissue in the body.
The gene defect also causes too much growth of the long bones of the body. This causes the tall height and long arms and legs seen in people with this syndrome. How this overgrowth happens is not well understood.
Other areas of the body that are affected include:
Symptoms: People with Marfan syndrome are usually tall with long, thin arms and legs and spider-like fingers -- called arachnodactyly. When they stretch out their arms, the length of their arms is greater than their height.
Other symptoms include:
Treatment:
Vision problems should be treated when possible.
Monitor for scoliosis, especially during the teenage years.
Medicine to slow the heart rate may help prevent stress on the aorta. Avoid participating in contact sports to avoid injuring the aorta of the heart. Some people may need surgery to replace the aortic root and valve.
People with Marfan syndrome who have heart valve conditions should take antibiotics before dental procedures to prevent endocarditis. Pregnant women with Marfan syndrome must be monitored very closely because of the increased stress on the heart and aorta.
Support Groups National Marfan Foundation -- www.marfan.org
Expectations (prognosis)Heart-related complications may shorten the lifespan of people with this disease. However, many patients survive well into their 60s. Good care and surgery may extend the lifespan further.
Complications may include:
Prevention Spontaneous new gene mutations leading to Marfan (less than 1/3 of cases) cannot be prevented. If you have Marfan syndrome, see your doctor at least once every year.
Disorders of connective tissue affect the skeletal system, cardiovascular system, eyes, and skin.
Causes, incidence, and risk factorsMarfan syndrome is caused by defects in a gene called fibrillin-1. Fibrillin-1 plays an important role as the building block for connective tissue in the body.
The gene defect also causes too much growth of the long bones of the body. This causes the tall height and long arms and legs seen in people with this syndrome. How this overgrowth happens is not well understood.
Other areas of the body that are affected include:
- Lung tissue (there may be a pneumothorax, in which air can escape from the lung into the chest cavity and collapse the lung)
- The aorta, the main blood vessel that takes blood from the heart to the body may stretch or become weak (called aortic dilation or aortic aneurysm)
- The eyes, causing cataracts and other problems (such as a dislocation of the lenses)
- The skin
- Tissue covering the spinal cord
Symptoms: People with Marfan syndrome are usually tall with long, thin arms and legs and spider-like fingers -- called arachnodactyly. When they stretch out their arms, the length of their arms is greater than their height.
Other symptoms include:
- A chest that sinks in or sticks out -- funnel chest (pectus excavatum) or pigeon breast (pectus carinatum)
- Flat feet
- Highly arched palate and crowded teeth
- Hypotonia
- Joints that are too flexible (but the elbows may be less flexible)
- Learning disability
- Movement of the lens of the eye from its normal position (dislocation)
- Nearsightedness
- Small lower jaw (micrognathia)
- Spine that curves to one side (scoliosis)
- Thin, narrow face
- Aneurysm
- Collapsed lung
- Heart valve problems
- Defects of the lens or cornea
- Retinal detachment
- Vision problems
- Echocardiogram
- Fibrillin-1 mutation testing (in some people)
Treatment:
Vision problems should be treated when possible.
Monitor for scoliosis, especially during the teenage years.
Medicine to slow the heart rate may help prevent stress on the aorta. Avoid participating in contact sports to avoid injuring the aorta of the heart. Some people may need surgery to replace the aortic root and valve.
People with Marfan syndrome who have heart valve conditions should take antibiotics before dental procedures to prevent endocarditis. Pregnant women with Marfan syndrome must be monitored very closely because of the increased stress on the heart and aorta.
Support Groups National Marfan Foundation -- www.marfan.org
Expectations (prognosis)Heart-related complications may shorten the lifespan of people with this disease. However, many patients survive well into their 60s. Good care and surgery may extend the lifespan further.
Complications may include:
- Aortic regurgitation
- Aortic rupture
- Bacterial endocarditis
- Dissecting aortic aneurysm
- Enlargement of the base of the aorta
- Heart failure
- Mitral valve prolapse
- Scoliosis
- Vision problems
Prevention Spontaneous new gene mutations leading to Marfan (less than 1/3 of cases) cannot be prevented. If you have Marfan syndrome, see your doctor at least once every year.
Edwards syndrome
Edwards syndrome is a chromosomal abnormality resulting in there being a third copy of chromosome 18 instead of the usual pair - this is called Trisomy 18.
Babies with Edwards Syndrome are likely to have some facially different (dysmorphic) features, as well as an increased risk of heart defects and difficulty with apnoea (remembering to breathe) in very young babies. Youngsters (particularly those with the most severe heart defects) are likely to need extensive medical support and due to the expected impairments this is frequently withheld in the mistaken belief that death is imminent and inevitable. It is not, and the oldest known person with the condition is in their early 30s at time of writing.
Infants that survive their first weeks and months will have physical and intellectual impairments but all will learn and develop with time, interact with others and experience a full range of emotions and pleasures. Approximately 1 in 10 babies will see their first birthday, and of these 1 in 10 will live to age 10.
Alport Syndrome
Alport Syndrome is an inherited disease that primarily affects the glomeruli, the tiny tufts of capillaries in the kidneys that filter wastes from the blood. The disease was first described by an English doctor named A. Cecil Alport. Alport Syndrome is caused by changes in genes (mutations) that affect type IV collagen, a protein that is important to the normal structure and function of glomeruli. The earliest symptom of the disease is blood in the urine (hematuria).
What are the symptoms? Alport Syndrome always affects the kidneys. Many people with Alport Syndrome also have hearing problems and abnormalities of the eyes, because the type IV collagen proteins are important to the normal structure and function of the inner ear and the eye..
Kidneys. The central feature of the disease is the presence of blood in the urine (hematuria). Boys with X-linked Alport Syndrome develop hematuria in infancy, and it is always present. The great majority of girls with X-linked Alport Syndrome also have hematuria, but it may come and go The hematuria of Alport Syndrome is usually microscopic, meaning it can only be detected with a microscope or a urine dipstick. Sometimes children with Alport Syndrome have brown, pink or red urine ( gross hematuria) for several days, brought on by a cold or the flu. This gross hematuria will go away on its own and while it may be frightening, it is not harmful.
As boys with Alport Syndrome grow, they begin to show other signs of kidney disease, including protein in the urine and high blood pressure. These symptoms are often present by the time the boys are teen-agers. Girls with Alport Syndrome usually do not have protein in the urine and high blood pressure until much later in life, but occasionally these symptoms appear in teen-aged girls with Alport Syndrome.
Ears. Deafness is another important feature of Alport Syndrome. About 80% of boys with Alport Syndrome will develop deafness at some point in their lives, often by the time they are teen-agers. The deafness affects both ears. Fortunately, hearing aids are usually very effective in these people. Girls with Alport Syndrome may also develop deafness, but less frequently than boys, and usually later in life. Kidney transplantation does not improve the deafness of Alport Syndrome.
Eyes. About 15% of men with Alport Syndrome have an abnormality in the shape of the lens called anterior lenticonus. People with anterior lenticonus may have some problems with their vision, and may develop cataracts.
How serious is Alport Syndrome? Alport Syndrome causes progressive kidney damage. The glomeruli and other normal kidney structures such as tubules are gradually replaced by scar tissue, leading to kidney failure. All boys with Alport Syndrome, regardless of the genetic type, eventually develop kidney failure. These boys often need dialysis or transplantation during their teen-age or young adult years, but kidney failure can occur as late as 40-50 years of age in some men with Alport Syndrome. Most girls with X-linked Alport Syndrome do not develop kidney failure. However, as woman with Alport Syndrome get older the possibility of kidney failure increases.
All boys and girls with autosomal recessive Alport Syndrome develop kidney failure, usually by their teens or young adult years. People with autosomal dominant Alport Syndrome are usually well into middle age before kidney failure develops.
How is Alport Syndrome diagnosed? Currently, diagnosis of Alport Syndrome relies on careful evaluation of the patient's clinical features, family history and results of tissue biopsies. Alport Syndrome produces unique changes in the walls of the blood vessels of the glomeruli that can be detected by electron microscopy of kidney biopsy material. Kidney biopsies can also be tested for the presence or absence of the type IV collagen alpha-3, alpha-4 and alpha-5 chains. This information is often very helpful in confirming a suspected diagnosis of Alport Syndrome. An alternative diagnostic procedure is skin biopsy. The type IV collagen alpha-5 chain is normally present in the skin. In most men with the X-linked form of Alport Syndrome the alpha-5 chain is completely missing from the skin.
How is Alport Syndrome treated? Currently there is no specific treatment for Alport Syndrome. The same treatments that are used in people with high blood pressure and other symptoms of kidney disease are used in people with Alport Syndrome. Kidney transplantation is usually very successful in people with Alport Syndrome, and is the best treatment when end-stage kidney failure is approaching.
Medical researchers are very interested in understanding why people with Alport Syndrome develop kidney failure, and in developing treatments that can slow or prevent the development of kidney failure. Several treatment approaches are being tested in animals with Alport Syndrome.
How is Alport Syndrome inherited? Type IV collagen is actually a family of six proteins, or chains, that are known as alpha-1, alpha-2, alpha-3, alpha-4, alpha-5 and alpha-6. Mutations that affect the alpha-3, alpha-4 or alpha-5 type IV collagen chains can cause Alport Syndrome
There are three genetic types of Alport Syndrome
X-linked. This is the most common form of Alport Syndrome. About 80% of the people with this disease have the X-linked type. Boys with this type are severely affected and always develop kidney failure sometime in their lives. Girls with this type usually have milder symptoms than boys, but they can develop kidney failure. The rest of the people with Alport Syndrome have either the autosomal recessive type which affects 15 percent, or the autosomal dominant type, which affects 5 percent.
What is the outlook for Alport Syndrome? Researchers recently isolated the gene responsible for Alport Syndrome. The research disclosed three mutations of the gene. This discovery not only will permit more precise diagnosis of Alport Syndrome, but it opens the possibility for future gene therapy for this disorder.
What are the symptoms? Alport Syndrome always affects the kidneys. Many people with Alport Syndrome also have hearing problems and abnormalities of the eyes, because the type IV collagen proteins are important to the normal structure and function of the inner ear and the eye..
Kidneys. The central feature of the disease is the presence of blood in the urine (hematuria). Boys with X-linked Alport Syndrome develop hematuria in infancy, and it is always present. The great majority of girls with X-linked Alport Syndrome also have hematuria, but it may come and go The hematuria of Alport Syndrome is usually microscopic, meaning it can only be detected with a microscope or a urine dipstick. Sometimes children with Alport Syndrome have brown, pink or red urine ( gross hematuria) for several days, brought on by a cold or the flu. This gross hematuria will go away on its own and while it may be frightening, it is not harmful.
As boys with Alport Syndrome grow, they begin to show other signs of kidney disease, including protein in the urine and high blood pressure. These symptoms are often present by the time the boys are teen-agers. Girls with Alport Syndrome usually do not have protein in the urine and high blood pressure until much later in life, but occasionally these symptoms appear in teen-aged girls with Alport Syndrome.
Ears. Deafness is another important feature of Alport Syndrome. About 80% of boys with Alport Syndrome will develop deafness at some point in their lives, often by the time they are teen-agers. The deafness affects both ears. Fortunately, hearing aids are usually very effective in these people. Girls with Alport Syndrome may also develop deafness, but less frequently than boys, and usually later in life. Kidney transplantation does not improve the deafness of Alport Syndrome.
Eyes. About 15% of men with Alport Syndrome have an abnormality in the shape of the lens called anterior lenticonus. People with anterior lenticonus may have some problems with their vision, and may develop cataracts.
How serious is Alport Syndrome? Alport Syndrome causes progressive kidney damage. The glomeruli and other normal kidney structures such as tubules are gradually replaced by scar tissue, leading to kidney failure. All boys with Alport Syndrome, regardless of the genetic type, eventually develop kidney failure. These boys often need dialysis or transplantation during their teen-age or young adult years, but kidney failure can occur as late as 40-50 years of age in some men with Alport Syndrome. Most girls with X-linked Alport Syndrome do not develop kidney failure. However, as woman with Alport Syndrome get older the possibility of kidney failure increases.
All boys and girls with autosomal recessive Alport Syndrome develop kidney failure, usually by their teens or young adult years. People with autosomal dominant Alport Syndrome are usually well into middle age before kidney failure develops.
How is Alport Syndrome diagnosed? Currently, diagnosis of Alport Syndrome relies on careful evaluation of the patient's clinical features, family history and results of tissue biopsies. Alport Syndrome produces unique changes in the walls of the blood vessels of the glomeruli that can be detected by electron microscopy of kidney biopsy material. Kidney biopsies can also be tested for the presence or absence of the type IV collagen alpha-3, alpha-4 and alpha-5 chains. This information is often very helpful in confirming a suspected diagnosis of Alport Syndrome. An alternative diagnostic procedure is skin biopsy. The type IV collagen alpha-5 chain is normally present in the skin. In most men with the X-linked form of Alport Syndrome the alpha-5 chain is completely missing from the skin.
How is Alport Syndrome treated? Currently there is no specific treatment for Alport Syndrome. The same treatments that are used in people with high blood pressure and other symptoms of kidney disease are used in people with Alport Syndrome. Kidney transplantation is usually very successful in people with Alport Syndrome, and is the best treatment when end-stage kidney failure is approaching.
Medical researchers are very interested in understanding why people with Alport Syndrome develop kidney failure, and in developing treatments that can slow or prevent the development of kidney failure. Several treatment approaches are being tested in animals with Alport Syndrome.
How is Alport Syndrome inherited? Type IV collagen is actually a family of six proteins, or chains, that are known as alpha-1, alpha-2, alpha-3, alpha-4, alpha-5 and alpha-6. Mutations that affect the alpha-3, alpha-4 or alpha-5 type IV collagen chains can cause Alport Syndrome
There are three genetic types of Alport Syndrome
X-linked. This is the most common form of Alport Syndrome. About 80% of the people with this disease have the X-linked type. Boys with this type are severely affected and always develop kidney failure sometime in their lives. Girls with this type usually have milder symptoms than boys, but they can develop kidney failure. The rest of the people with Alport Syndrome have either the autosomal recessive type which affects 15 percent, or the autosomal dominant type, which affects 5 percent.
What is the outlook for Alport Syndrome? Researchers recently isolated the gene responsible for Alport Syndrome. The research disclosed three mutations of the gene. This discovery not only will permit more precise diagnosis of Alport Syndrome, but it opens the possibility for future gene therapy for this disorder.
Riley-Day syndrome
Riley-Day syndrome is an inherited disorder that affects the development and function of nerves throughout the body.
CausesRiley-Day syndrome is passed down through families (inherited). A person must inherit a copy of the defective gene from each parent to develop the condition.
This condition is seen most often in people of Eastern European Jewish ancestry (Ashkenazi Jews), where the incidence is 1 in 3,700. The disease is caused by a change (mutation) of the IKBKAP gene on chromosome 9. It is rare in the general population.
Symptoms
Symptoms are present at birth and grow worse over time.
Exams and Tests The health care provider will perform a physical exam. The patient may have:
Treatment may include:
Possible Complications The following complications occur in about 40% of patients with this condition:
PreventionPeople who are of an Eastern European Jewish background and families with a history of Riley-Day syndrome who are thinking of having children can seek genetic counseling to discuss their risk and undergo testing, when appropriate.
Genetic testing by DNA is very accurate for Riley-Day syndrome. It may be used for diagnosing affected individuals, detecting carriers, and prenatal diagnosis.
CausesRiley-Day syndrome is passed down through families (inherited). A person must inherit a copy of the defective gene from each parent to develop the condition.
This condition is seen most often in people of Eastern European Jewish ancestry (Ashkenazi Jews), where the incidence is 1 in 3,700. The disease is caused by a change (mutation) of the IKBKAP gene on chromosome 9. It is rare in the general population.
Symptoms
- Breath holding spells (can lose consciousness)
- Constipation
- Decreased taste
- Diarrhea
- Dry eyes
- Feeding difficulties
- Inability to feel pain and changes in temperature (can lead to injuries)
- Lack of tears when emotional crying
- Long episodes of vomiting
- Poor coordination - unsteady walk
- Poor growth
- Repeated fevers
- Repeated pneumonia
- Seizures
- Skin blotching
- Sweating while eating
- Unusually smooth, pale tongue surface
Symptoms are present at birth and grow worse over time.
Exams and Tests The health care provider will perform a physical exam. The patient may have:
- Absent or decreased deep tendon reflexes
- Lack of a response after receiving a histamine injection (normally redness and swelling would occur)
- Lack of tears with emotional crying
- Low muscle tone (hypotonia), especially in babies
- Repeated episodes of high blood pressure
- Severe scoliosis
- Tiny pupils after receiving certain eye drops
Treatment may include:
- Anticonvulsant therapy if there are seizures
- Feeding in an upright position and giving textured formula to prevent gastroesophageal reflux
- Increased fluid and salt intake, caffeine, and elastic stockings to prevent low blood pressure when standing (postural hypotension)
- Medicines called antiemetics, to control vomiting
- Medicines, including liquid tears, to prevent dry eyes
- Physical therapy of the chest
- Protecting the person from injury
- Providing enough nutrition and fluids
- Surgery or spinal fusion
- Treating aspiration pneumonia
Possible Complications The following complications occur in about 40% of patients with this condition:
- Blotching of the face and torso
- Excessive sweating of the head and torso
- High blood pressure (hypertension) and rapid heart rate (tachycardia)
- Insomnia
- Irritability
- Mottling of the hands and feet
- Nausea/vomiting
- Severe difficulty swallowing (dysphagia), drooling
- Worsening of muscle tone
PreventionPeople who are of an Eastern European Jewish background and families with a history of Riley-Day syndrome who are thinking of having children can seek genetic counseling to discuss their risk and undergo testing, when appropriate.
Genetic testing by DNA is very accurate for Riley-Day syndrome. It may be used for diagnosing affected individuals, detecting carriers, and prenatal diagnosis.
Horner Syndrome
Horner syndrome is a rare condition that affects the nerves to the eye and face.
Causes Horner syndrome can be caused by any interruption in a set of nerve fibers that start in the part of the brain called the hypothalamus and travel to the face and eyes.
Sympathetic nerve fiber injuries can result from:
Symptoms
Exams and Tests An eye examination may show:
Other tests may include:
Treatment depends on the cause of the problem. There is no treatment for Horner syndrome itself.
Outlook (Prognosis)The outcome depends on whether treatment of the cause is successful.
Possible ComplicationsThere are no direct complications of Horner syndrome itself. However, there may be complications from the disease that caused Horner syndrome or from its treatment.
When to Contact a Medical Professional Call your health care provider if you have symptoms of Horner syndrome.
Causes Horner syndrome can be caused by any interruption in a set of nerve fibers that start in the part of the brain called the hypothalamus and travel to the face and eyes.
Sympathetic nerve fiber injuries can result from:
- Injury to one of the main arteries to the brain (carotid artery)
- Injury to nerves at the base of the neck called the brachial plexus
- Migraine or cluster headaches
- Stroke, tumor, or other damage to a part of the brain called the brainstem
- Tumor in the top of the lung
Symptoms
- Decreased sweating on the affected side of the face
- Drooping eyelid (ptosis)
- Sinking of the eyeball into the face
- Small (constricted) pupil (the black part in the center of the eye)
Exams and Tests An eye examination may show:
- Changes in how the pupil opens or closes
- Eyelid drooping
- Red eye
Other tests may include:
- Blood tests
- Blood vessel tests (cerebral angiogram, CT angiogram, or MR angiogram)
- Chest x-ray or chest CT scan
- MRI or CT scan of the brain
- Spinal tap (lumbar puncture)
Treatment depends on the cause of the problem. There is no treatment for Horner syndrome itself.
Outlook (Prognosis)The outcome depends on whether treatment of the cause is successful.
Possible ComplicationsThere are no direct complications of Horner syndrome itself. However, there may be complications from the disease that caused Horner syndrome or from its treatment.
When to Contact a Medical Professional Call your health care provider if you have symptoms of Horner syndrome.
Thalassemia
If you have thalassemia, your body has problems making hemoglobin, the protein in red blood cells that carries oxygen through your body. When your blood does not carry enough oxygen to the rest of your body, you have anemia.
Thalassemia, a genetic disease, can be mild or severe. Some carriers of the gene have no symptoms. The most common severe form in the United States is a type called Cooley's anemia. It mainly affects people of Mediterranean or Asian ancestry. It usually appears during the first two years of life. Severe thalassemia is treated with blood transfusions and treatment to remove excess iron in the blood.
Thalassemia, a genetic disease, can be mild or severe. Some carriers of the gene have no symptoms. The most common severe form in the United States is a type called Cooley's anemia. It mainly affects people of Mediterranean or Asian ancestry. It usually appears during the first two years of life. Severe thalassemia is treated with blood transfusions and treatment to remove excess iron in the blood.
Klinefelter's Syndrome
Klinefelter's syndrome is a condition that occurs in men who have an extra X chromosome in most of their cells. The syndrome can affect different stages of physical, language and social development. The most common symptom is infertility. Because they often don't make as much of the male hormone testosterone as other boys, teenagers with Klinefelter's syndrome may have less facial and body hair and may be less muscular than other boys. They may have trouble using language to express themselves. They may be shy and have trouble fitting in.
It is important to start treatment as early as possible. With treatment, most boys grow up to have normal sex lives, successful careers and normal social relationships. Treatments include
It is important to start treatment as early as possible. With treatment, most boys grow up to have normal sex lives, successful careers and normal social relationships. Treatments include
- Educational services
- Physical, speech and occupational therapy
- Medical treatments including testosterone replacement
